Fundamento y Antecedentes
El genotipado basado en secuencias (SBG), por sus siglas en ingles "Sequence Based Genotyping", incluye las técnicas para el estudio de la diversidad genética utilizando las tecnológías de Secuenciación de Nueva Generación (por sus siglas en inglés NGS, Next Generation Sequencing) que buscan la caracterización genética de los organismo en base a la generación de datos de decenas o incluso cientos de miles de marcadores moleculares, por medio de la secuencición másiva de ADN.
Se han desarrollado varias técnicas de SBG, entre las que se encuentra El Genotipado por Secuenciación GBS (Genotyping By Sequencing); tecnología de matriz de diversidad DArTseq, método desarrollado por la empresa Diversity Arrays Technology (DArT); tecnología de secuenciación de DNA asociadas a sitios de restricción RAD-Seq, por sus siglas en ingles "Restriction site Associated DNA Sequencing", pudiendo ser está de doble digestión (ddRAD-Seq), entre otros.
La secuenciación masiva puede ser aplicada a bibliotecas genómicas que consigan una reducción del genoma de los organismos y a partir de la cual se obtenga un número elevado de variantes genómicas en múltiples individuos. La reducción del genoma se consigue con estas metodologías mediante el empleo de endonucleasas de restricción, que son enzimas que reconocen un patrón específico de bases en las secuencias de ADN.
Las bibliotecas de ADN obtenidas con estas metodológias y los enfoques de genotipado por secuenciación, aprovechan las ventajas de las plataformas de secuenciación de nueva generación (NGS) para crear lecturas cortas de miles de regiones potencialmente homólogas a través de múltiples individuos, dirigiéndose a regiones genómicas adyacentes a sitios de corte de enzimas de restricción. En contraste con los datos "shotgun" del genoma completo, estos proporcionan una fuente más eficiente y económica para realizar comparaciones entre individuos de la misma especie, lo que la convierte en una herramienta popular para los análisis genéticos de poblaciones.
Las lecturas obtenidas por estas metodologías se pueden usar para la detección y genotipado del polimorfismo sin el uso de un genoma de referencia, por lo tanto, permiten la genotipificación en prácticamente cualquier organismo o especie.

Muestra los métodos de genotipado denominados ddRAD-Seq, optimizados y flexibles para el número de marcadores genéticos con diferente enfoque experimental específico para un sistema biológico determinado.
Servicio ddRAD-Seq
LabSerGen cuenta con la plataforma de Secuenciación y análisis para ddRAD-Seq, utilizando el corte dual por las enzimas de restricción 'Bgl-II' y 'Dde-I' (ddRAD-Seq), y adaptadores complementarios a la plataforma de Illumina, esta metodología está basado en la modificación al protocolo de Brant K. Peterson.
Se cuenta con las capacidades para los análisis bioinformáticos para el entendimiento e interpretación de los RAD-Seq, el cual incluye diversidad genética, arboles filogénicos y análisis de componentes, de la genética poblacional.
En caso de contar con genóma de referencia, es posible evaluar el número de lecturas, así como el tamaño de estas, realizando una simulación in silicon de los cortes de las enzímas de restricción.
La referencia de forma habitual es tomada de la Base de Datos de NCBI.
Simulación in silicon de la referencia para evaluar tamaño de las lecturas y profundidad de análisis.

Fig. 2 La grafica indica el número de lecturas para una profundidad o cobertura indicada en el eje Y.


Análisis Bioinformático
Nota: En caso de solicitar el servicio de análisis bioinformático, este se limita a identificar la huella genética para cada individuo (SNPs y/o suma de SNPs), comparación entre individuos, agrupamiento para identificar las poblaciones, análisis filogénico y análisis de componente para evaluar la carga genética de cada individuo o muestra.
Flujo de Trabajo
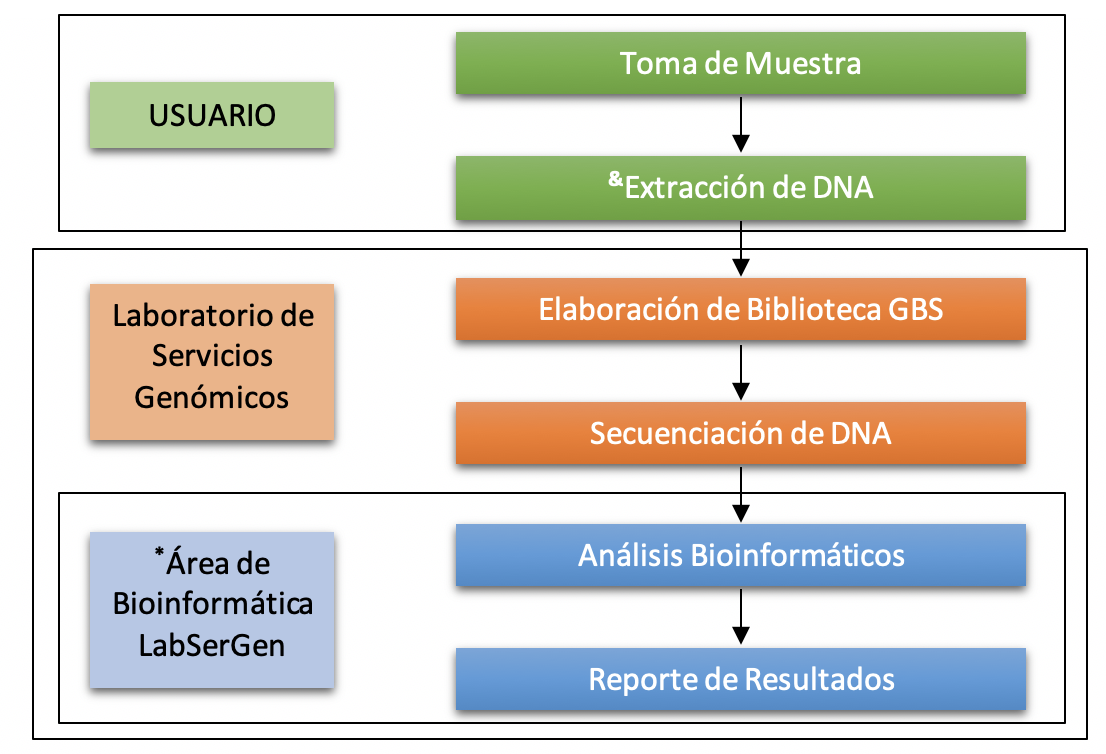
Nota: Si requiere servicio de Extracción de ADN, contacte al laboratorio, el servicio tiene un costo extra.
Cualquier análisis realizado por el Área de Bioinformática del Laboratorio de Servicios Genómicos tiene un costo adicional.
Nota: El laboratorio cuenta con los servicios de análisis bioinformático.
Nota: El laboratorio cuenta con los servicios de extracción de DNA.
Requisitos de la muestra
-
El usuario deberá definir el número de reads requeridos, tamaño de los insertos a secuenciar, porcentaje del genoma a secuenciar, la profundidad y formato de corrida.
-
Entregar DNA genómico de 0.5 a 1 μg, diluido en agua, integro, limpio, y a una concentración no menor de 50 ng/μl o puede enviarse la muestra liofilizada.
-
El usuario deberá proveer evidencia de que el cociente DO 260/280 es de por lo menos 1.8.
-
Incluir fotografía o gráfica que muestre la calidad e integridad de la muestra.
Nota: Cualquier contaminación en la muestra se reflejará en la calidad y/o rendimiento de los resultados o bien puede inhibir las reacciones iniciales del proceso lo que no permitiría obtener la biblioteca.
Entrega de Resultados
Entrega de Resultados del proceso de Secuenciación:
-
Reporte en formato PDF, indicando el proceso que se siguió con las muestras con sus respectivo Identificadores.
-
Archivo en formato fastq. incluye las lecturas (reads) y sus calidades definidas en base al estándar de calidad Phred, con un promedio de calidad arriba de 20.
Entrega de Resultados del proceso de Análisis Bioinformático (en caso de ser solicitado):
-
Reporte en formato PDF, el cual indica el proceso de análisis y resumen de resultados.
-
Análisis y Filtrado de Calidad.
-
Agrupamiento, alineación, ensamblado y formación de clusters, análisis de novo.
-
Generación de datos de salida (archivos): .vcf, .loci, .phy, .snps, .nex, .snps.geno, .ped, .map y .fasta
-
Análisis genómicos terciario: construcción del árbol filogenético